
Chapter 13: Thermodynamics
13.4 Free Energy
Learning Outcomes
- Define Gibbs free energy, and describe its relation to spontaneity
- Explain how temperature affects the spontaneity of some processes and predict spontaneity from [latex]\Delta[/latex]H and [latex]\Delta[/latex]S
- Calculate free energy change for:
- a process using enthalpies of formation and the entropies for its reactants and products
- a reaction using free energies of formation for its reactants and products
- coupled reactions
One of the challenges of using the second law of thermodynamics to determine if a process is spontaneous is that it requires measurements of the entropy change for the system and the entropy change for the surroundings. An alternative approach involving a new thermodynamic property defined in terms of system properties only was introduced in the late nineteenth century by American mathematician Josiah Willard Gibbs. This new property is called the Gibbs free energy (G) (or simply the free energy), and it is defined in terms of a system’s enthalpy and entropy as the following:
G = H – TS
Free energy is a state function, and at constant temperature and pressure, the standard free energy change ([latex]\Delta[/latex]G°) may be expressed as the following:
[latex]\Delta[/latex]G° = [latex]\Delta[/latex]H – T[latex]\Delta[/latex]S
(For simplicity’s sake, the subscript “sys” will be omitted henceforth.)
We can understand the relationship between this system property and the spontaneity of a process by recalling the previously derived second law expression:
[latex]\Delta {S}_{\text{univ}}=\Delta S+\dfrac{{q}_{\text{surr}}}{T}[/latex]
The first law requires that qsurr = −qsys, and at constant pressure qsys = [latex]\Delta[/latex]H, and so this expression may be rewritten as the following:
[latex]\Delta {S}_{\text{univ}}=\Delta S-\dfrac{\Delta H}{T}[/latex]
[latex]\Delta[/latex]H is the enthalpy change of the system. Multiplying both sides of this equation by −T, and rearranging yields the following:
–T[latex]\Delta[/latex]Suniv = [latex]\Delta[/latex]H – T[latex]\Delta[/latex]S
Comparing this equation to the previous one for free energy change shows the following relation:
[latex]\Delta[/latex]G° = –T[latex]\Delta[/latex]Suniv
The free energy change is therefore a reliable indicator of the spontaneity of a process, being directly related to the previously identified spontaneity indicator, [latex]\Delta[/latex]Suniv. Table 13.4.1 summarizes the relation between the spontaneity of a process and the arithmetic signs of these indicators.
What’s “Free” about [latex]\Delta[/latex]G?
In addition to indicating spontaneity, the free energy change also provides information regarding the amount of useful work (w) that may be accomplished by a spontaneous process. Although a rigorous treatment of this subject is beyond the scope of an introductory chemistry text, a brief discussion is helpful for gaining a better perspective on this important thermodynamic property.
For this purpose, consider a spontaneous, exothermic process that involves a decrease in entropy. The free energy, as defined by
[latex]\Delta[/latex]G° = [latex]\Delta[/latex]H – T[latex]\Delta[/latex]S
may be interpreted as representing the difference between the energy produced by the process, [latex]\Delta[/latex]H, and the energy lost to the surroundings, T[latex]\Delta[/latex]S. The difference between the energy produced and the energy lost is the energy available (or “free”) to do useful work by the process, [latex]\Delta[/latex]G. If the process somehow could be made to take place under conditions of thermodynamic reversibility, the amount of work that could be done would be maximal:
[latex]\Delta[/latex]G = wmax
However, as noted previously in this chapter, such conditions are not realistic. In addition, the technologies used to extract work from a spontaneous process (e.g., automobile engine, steam turbine) are never 100% efficient, and so the work done by these processes is always less than the theoretical maximum. Similar reasoning may be applied to a nonspontaneous process, for which the free energy change represents the minimum amount of work that must be done on the system to carry out the process.
Calculating Free Energy Change
Free energy is a state function, so its value depends only on the conditions of the initial and final states of the system that have undergone some change. A convenient and common approach to the calculation of free energy changes for physical and chemical reactions is by use of widely available compilations of standard state thermodynamic data. One method involves the use of standard enthalpies and entropies to compute standard free energy changes according to the following relation as demonstrated in Example 1.
[latex]\Delta[/latex] G° = [latex]\Delta[/latex] H° – T[latex]\Delta[/latex]S°
Example 13.4.1: Evaluation of [latex]\Delta[/latex]G° Change from [latex]\Delta[/latex]H° and [latex]\Delta[/latex]S°
Use standard enthalpy and entropy data from Standard Thermodynamic Properties for Selected Substances to calculate the standard free energy change for the vaporization of water at room temperature (298 K). What does the computed value for [latex]\Delta[/latex]G° say about the spontaneity of this process?
Show Solution
The process of interest is the following:
[latex]\ce{H2O}\left(l\right)\longrightarrow \ce{H2O}\left(g\right)[/latex]
The standard change in free energy may be calculated using the following equation:
[latex]\Delta {G}^{\circ }=\Delta H^{\circ}-T\Delta S^{\circ}[/latex]
From Standard Thermodynamic Properties for Selected Substances, here is the data:
| Substance | [latex]\Delta {H}_{\text{f}}^{\circ }\text{(kJ/mol)}[/latex] | [latex]{S}^{\circ }\text{(J/K} \cdot \text{mol)}[/latex] |
|---|---|---|
| [latex]\ce{H2O}[/latex](l) | −285.83 | 70.0 |
| [latex]\ce{H2O}[/latex](g) | −241.82 | 188.8 |
Using the appendix data to calculate the standard enthalpy and entropy changes yields:
[latex]\begin{array}{rll}\Delta H^{\circ }&=&\Delta {H}^{\circ }=\Delta {H}_{\text{f}}^{\circ }\left(\ce{H2O}\left(g\right)\right)-\Delta {H}_{\text{f}}^{\circ }\left(\ce{H2O}\left(l\right)\right)\\{}&=&\left[-\text{241.82 kJ}-\left(-\text{285.83}\right)\right]\text{kJ/mol}=\text{44.01 kJ/mol}\end{array}[/latex]
[latex]\begin{array}{rll}\Delta S^{\circ }&=&\Delta {S}^{\circ }={S}^{\circ }\left(\ce{H2O}\left(g\right)\right)-{S}^{\circ }\left(\ce{H2O}\left(l\right)\right)\\{}&=&188.8\text{J/mol}\cdot\text{K}-70.0\text{J/K}=118.8\text{J/mol}\cdot\text{K}\end{array}[/latex]
[latex]\Delta G^{\circ }=\Delta H^{\circ }-T\Delta S^{\circ }[/latex]
Substitution into the standard free energy equation yields:
[latex]\begin{array}{rll}\Delta {G}^{\circ }&=&\Delta H^{\circ }-T\Delta S^{\circ }\\{}&=&\text{44.01 kJ/mol}-\left(\text{298 K}\times 118.8\text{J/mol}\cdot\text{K}\right)\times \dfrac{\text{1 kJ}}{\text{1000 J}}\end{array}[/latex]
[latex]\text{44.01 kJ/mol}-\text{35.4 kJ/mol}=\text{8.6 kJ/mol}[/latex]
At 298 K (25 °C) [latex]\Delta {G}^{\circ }>0[/latex], and so boiling is nonspontaneous (not spontaneous).
Check Your Learning
The standard free energy change for a reaction may also be calculated from standard free energy of formation [latex]\left(\Delta {G}_{\text{f}}^{\circ }\right)[/latex] values of the reactants and products involved in the reaction. The standard free energy of formation is the free energy change that accompanies the formation of one mole of a substance from its elements in their standard states. Similar to the standard enthalpies of formation, [latex]\Delta {G}_{\text{f}}^{\circ }[/latex] is by definition zero for elemental substances under standard state conditions. The approach to computing the free energy change for a reaction using this approach is the same as that demonstrated previously for enthalpy and entropy changes. For the reaction
[latex]m\text{A}+n\text{B}\longrightarrow x\text{C}+y\text{D}[/latex],
the standard free energy change at room temperature may be calculated as
[latex]\begin{array}{rll}{}\Delta {G}^{\circ }=\Delta G^{\circ }&=&\sum \nu \Delta {G}^{\circ }\left(\text{products}\right)-\sum \nu \Delta {G}^{\circ }\left(\text{reactants}\right)\\{}&=&\left[x\Delta {G}_{\text{f}}^{\circ }\left(\text{C}\right)+y\Delta {G}_{\text{f}}^{\circ }\left(\text{D}\right)\right]-\left[m\Delta {G}_{\text{f}}^{\circ }\left(\text{A}\right)+n\Delta {G}_{\text{f}}^{\circ }\left(\text{B}\right)\right]\end{array}[/latex]
Example 13.4.2: Calculation of [latex]\Delta {G}_{298}^{\circ }[/latex]
Consider the decomposition of yellow mercury(II) oxide.
[latex]\ce{HgO}\left(s,\text{yellow}\right)\longrightarrow \ce{Hg}\left(l\right)+\frac{1}{2}\ce{O2}\left(g\right)[/latex]
Calculate the standard free energy change at room temperature, [latex]\Delta {G}_{298}^{\circ }[/latex], using (a) standard free energies of formation and (b) standard enthalpies of formation and standard entropies. Do the results indicate the reaction to be spontaneous or nonspontaneous under standard conditions?
Show Solution
The required data are available in Standard Thermodynamic Properties for Selected Substances and are shown here.
| Compound | [latex]\Delta {G}_{\text{f}}^{\circ }\text{(kJ/mol)}[/latex] | [latex]\Delta {H}_{\text{f}}^{\circ }\text{(kJ/mol)}[/latex] | [latex]{S}_{298}^{\circ }\text{(J/K}\cdot\text{mol)}[/latex] |
|---|---|---|---|
| [latex]\ce{HgO}[/latex] (s, yellow) | −58.43 | −90.46 | 71.13 |
| [latex]\ce{Hg}[/latex](l) | 0 | 0 | 75.9 |
| [latex]\ce{O2}[/latex](g) | 0 | 0 | 205.2 |
- Using free energies of formation:
[latex]\begin{array}{rll}{}\Delta {G}^{\circ }&=&\sum \nu G_{\text{f}}^{\circ }\text{(products)}-\sum \nu \Delta {G}_{\text{f}}^{\circ }\text{(reactants)}\\{}&=&\left[1\Delta {G}_{\text{f}}^{\circ }\text{Hg}\left(l\right)+\frac{1}{2}\Delta {G}_{\text{f}}^{\circ }{\text{O}}_{\text{2}}\left(g\right)\right]-1\Delta {G}_{\text{f}}^{\circ }\text{HgO}\left(s,\text{yellow}\right)\\{}&=&\left[1\text{mol}\text{(0 kJ/mol)}+\frac{1}{2}\text{mol(0 kJ/mol)}\right]-\text{1 mol(-58.43 kJ/mol)}=\text{58.43 kJ/mol}\end{array}[/latex] - Using enthalpies and entropies of formation:
[latex]\begin{array}{rll}{}\Delta {H}^{\circ }&=&\sum \nu \Delta {H}_{\text{f}}^{\circ }\text{(products)}-\sum \nu \Delta {H}_{\text{f}}^{\circ }\text{(reactants)}\\{}&=&\left[1\Delta {H}_{\text{f}}^{\circ }\text{Hg}\left(l\right)+\frac{1}{2}\Delta {H}_{\text{f}}^{\circ }{\ce{O}}_{2}\left(g\right)\right]-1\Delta {H}_{\text{f}}^{\circ }\ce{HgO}\left(s,\text{yellow}\right)\\{}&=&\left[\text{1 mol}\left(\text{0 kJ/mol}\right)+\frac{1}{2}\text{mol}\left(\text{0 kJ/mol}\right)\right]-\text{1 mol}\left(-\text{90.46 kJ/mol}\right)=\text{90.46 kJ/mol}\end{array}[/latex]
[latex]\begin{array}{rll}\\ \Delta {S}^{\circ }&=&\sum\nu\Delta {S}^{\circ }\text{(products)}-\sum \nu\Delta {S}^{\circ }\text{(reactants)}\\{}&=&\left[1\Delta {S}^{\circ }\text{Hg}\left(l\right)+\frac{1}{2}\Delta {S}^{\circ }{\ce{O}}_{2}\left(g\right)\right]-1\Delta {S}^{\circ }\ce{HgO}\left(s,\text{yellow}\right)\\{}&=&\left[\text{1 mol}\left(\text{75.9 J/mol K}\right)+\frac{1}{2}\text{mol}\left(\text{205.2 J/mol K}\right)\right]-\text{1 mol}\left(\text{71.13 J/mol K}\right)=\text{107.4 J/mol K}\end{array}[/latex]
[latex]\begin{array}{rll}\\ \Delta G^{\circ }&=&\Delta H^{\circ }-T\Delta S^{\circ }=\text{90.46 kJ}-\text{298.15 K}\times \text{107.4 J/K}\cdot \text{mol}\times \dfrac{\text{1 kJ}}{\text{1000 J}}\\\Delta G^{\circ }&=&\left(90.46 - 32.01\right)\text{kJ/mol}=\text{58.45 kJ/mol}\end{array}[/latex]
Both ways to calculate the standard free energy change at 25 °C give the same numerical value (to three significant figures), and both predict that the process is nonspontaneous (not spontaneous) at room temperature.
Check Your Learning
Free Energy Changes for Coupled Reactions
The use of free energies of formation to compute free energy changes for reactions as described above is possible because [latex]\Delta[/latex]G is a state function, and the approach is analogous to the use of Hess’ Law in computing enthalpy changes (see the chapter on thermochemistry). Consider the vaporization of water as an example:
[latex]\ce{H2O} (l) \rightarrow \ce{H2O} (g)[/latex]
An equation representing this process may be derived by adding the formation reactions for the two phases of water (necessarily reversing the reaction for the liquid phase). The free energy change for the sum reaction is the sum of free energy changes for the two added reactions:
[latex]\ce{H2} (g) + \frac{1}{2} \ce{O2} (g) \rightarrow \ce{H2O} (g)[/latex] [latex]\Delta G_f^\circ \text{gas}[/latex]
[latex]\ce{H2O} (l) \rightarrow \ce{H2} (g) + \frac{1}{2} \ce{O2} (g)[/latex] [latex]-\Delta G_f^\circ \text{liquid}[/latex]
[latex]\ce{H2O} (l) \rightarrow \ce{H2O} (g)[/latex] [latex]\Delta G^\circ = \Delta G_f^\circ \text{gas} -\Delta G_f^\circ \text{liquid}[/latex]
This approach may also be used in cases where a nonspontaneous reaction is enabled by coupling it to a spontaneous reaction. For example, the production of elemental zinc from zinc sulfide is thermodynamically unfavorable, as indicated by a positive value for [latex]\Delta[/latex]G°:
[latex]\ce{ZnS} (s) \rightarrow \ce{Zn} (s) + \ce{S} (s)[/latex] [latex]\Delta G_1^\circ = 201.3 \text{kJ}[/latex]
The industrial process for production of zinc from sulfidic ores involves coupling this decomposition reaction to the thermodynamically favorable oxidation of sulfur:
[latex]\ce{S} (s) + \ce{O2}(g) \rightarrow \ce{SO2} (g)[/latex] [latex]\Delta G_2^\circ = -300.1\text{kJ}[/latex]
The coupled reaction exhibits a negative free energy change and is spontaneous:
[latex]\ce{ZnS} (s) + \ce{O2}(g) \rightarrow \ce{Zn} (s) + \ce{SO2} (g)[/latex] [latex]\Delta G^\circ = 201.3 \text{kJ} + -300.1 \text{kJ} = -98.8 \text{kJ}[/latex]
This process is typically carried out at elevated temperatures, so this result obtained using standard free energy values is just an estimate. The gist of the calculation, however, holds true.
Example 13.4.3: Calculating Free Energy Change for a Coupled Reaction
Is a reaction coupling the decomposition of [latex]\ce{ZnS}[/latex] to the formation of H2S expected to be spontaneous under standard conditions?
Show Solution
Following the approach outlined above and using free energy values from Standard Thermodynamic Properties for Selected Substances.
[latex]\text{Decomposition of zinc sulfide:}[/latex] [latex]\ce{ZnS} (s) \rightarrow \ce{Zn} (s) + \ce{S} (s)[/latex] [latex]\Delta G_1^\circ = 201.3 \text{kJ}[/latex]
[latex]\text{Formation of hydrogen sulfide:}[/latex] [latex]\ce{S} (s) + \ce{H2} (g) \rightarrow \ce{H2}\text{S} (g)[/latex] [latex]\Delta G_2^\circ = -33.4 \text{kJ}[/latex]
[latex]\text{Coupled reaction:}[/latex] [latex]\ce{ZnS} (s) + \ce{H2} (g) \rightarrow \ce{Zn} (s) +\ce{H2S}}(g)[/latex] [latex]\Delta G^\circ = 201.3\text{kJ} + -33.4 \text{kJ} = 167.9 \text{kJ}[/latex]
The coupled reaction exhibits a positive free energy change and is thus nonspontaneous.
Check Your Learning
Temperature Dependence of Spontaneity
As was previously demonstrated in this chapter’s section on entropy, the spontaneity of a process may depend upon the temperature of the system. Phase transitions, for example, will proceed spontaneously in one direction or the other depending upon the temperature of the substance in question. Likewise, some chemical reactions can also exhibit temperature dependent spontaneities. To illustrate this concept, the equation relating free energy change to the enthalpy and entropy changes for the process is considered:
[latex]\Delta G=\Delta H-T\Delta S[/latex]
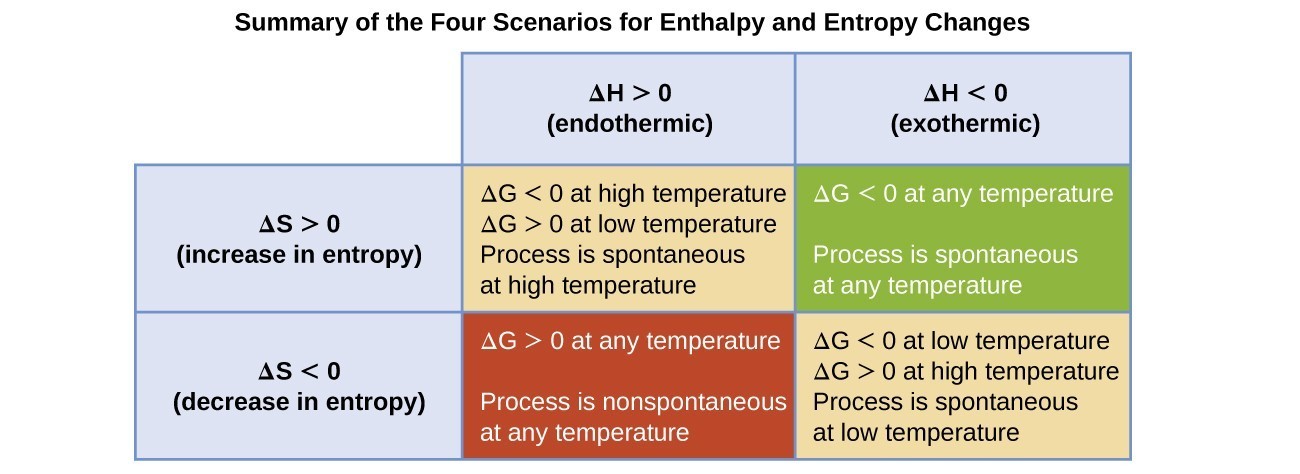
The spontaneity of a process, as reflected in the arithmetic sign of its free energy change, is then determined by the signs of the enthalpy and entropy changes and, in some cases, the absolute temperature. Since T is the absolute (kelvin) temperature, it can only have positive values. Four possibilities therefore exist with regard to the signs of the enthalpy and entropy changes:
- Both [latex]\Delta[/latex]H and [latex]\Delta[/latex]S are positive. This condition describes an endothermic process that involves an increase in system entropy. In this case, [latex]\Delta[/latex]G will be negative if the magnitude of the T[latex]\Delta[/latex]S term is greater than [latex]\Delta[/latex]H. If the T[latex]\Delta[/latex]S term is less than [latex]\Delta[/latex]H, the free energy change will be positive. Such a process is spontaneous at high temperatures and nonspontaneous at low temperatures.
- Both [latex]\Delta[/latex]H and [latex]\Delta[/latex]S are negative. This condition describes an exothermic process that involves a decrease in system entropy. In this case, [latex]\Delta[/latex]G will be negative if the magnitude of the T[latex]\Delta[/latex]S term is less than [latex]\Delta[/latex]H. If the T[latex]\Delta[/latex]S term’s magnitude is greater than [latex]\Delta[/latex]H, the free energy change will be positive. Such a process is spontaneous at low temperatures and nonspontaneous at high temperatures.
- [latex]\Delta[/latex]H is positive and [latex]\Delta[/latex]S is negative. This condition describes an endothermic process that involves a decrease in system entropy. In this case, [latex]\Delta[/latex]G will be positive regardless of the temperature. Such a process is nonspontaneous at all temperatures.
- [latex]\Delta[/latex]H is negative and [latex]\Delta[/latex]S is positive. This condition describes an exothermic process that involves an increase in system entropy. In this case, [latex]\Delta[/latex]G will be negative regardless of the temperature. Such a process is spontaneous at all temperatures.
These four scenarios are summarized in Figure 13.4.1.
Example 13.4.4: Predicting the Temperature Dependence of Spontaneity
The incomplete combustion of carbon is described by the following equation:
[latex]\ce{2C}\left(s\right)+\ce{O2}\left(g\right)\longrightarrow \ce{2CO}\left(g\right)[/latex]
How does the spontaneity of this process depend upon temperature?
Show Solution
Combustion processes are exothermic ([latex]\Delta[/latex]H < 0). This particular reaction involves an increase in entropy due to the accompanying increase in the amount of gaseous species (net gain of one mole of gas, [latex]\Delta[/latex]S > 0). The reaction is therefore spontaneous ([latex]\Delta[/latex]G < 0) at all temperatures.
Check Your Learning
When considering the conclusions drawn regarding the temperature dependence of spontaneity, it is important to keep in mind what the terms “high” and “low” mean. Since these terms are adjectives, the temperatures in question are deemed high or low relative to some reference temperature. A process that is nonspontaneous at one temperature but spontaneous at another will necessarily undergo a change in “spontaneity” (as reflected by its [latex]\Delta[/latex]G) as temperature varies. This is clearly illustrated by a graphical presentation of the free energy change equation, in which [latex]\Delta[/latex]G is plotted on the y axis versus T on the x axis:
[latex]\Delta G=\Delta H-T\Delta S[/latex]
[latex]y=b+mx[/latex]
Such a plot is shown in Figure 13.4.2. A process whose enthalpy and entropy changes are of the same arithmetic sign will exhibit a temperature-dependent spontaneity as depicted by the two yellow lines in the plot. Each line crosses from one spontaneity domain (positive or negative [latex]\Delta[/latex]G) to the other at a temperature that is characteristic of the process in question. This temperature is represented by the x-intercept of the line, that is, the value of T for which [latex]\Delta[/latex]G is zero:
[latex]\Delta G=0=\Delta H-T\Delta S[/latex]
[latex]T=\dfrac{\Delta H}{\Delta S}[/latex]
And so, saying a process is spontaneous at “high” or “low” temperatures means the temperature is above or below, respectively, that temperature at which [latex]\Delta[/latex]G for the process is zero. As noted earlier, this condition describes a system at equilibrium.
Example 13.5.5: Equilibrium Temperature for a Phase Transition
As defined in the chapter on liquids and solids, the boiling point of a liquid is the temperature at which its solid and liquid phases are in equilibrium (that is, when vaporization and condensation occur at equal rates). Use the information in Standard Thermodynamic Properties for Selected Substances to estimate the boiling point of water.
Show Solution
The process of interest is the following phase change:
[latex]\ce{H2O}\left(l\right)\longrightarrow \ce{H2O}\left(g\right)[/latex]
When this process is at equilibrium, [latex]\Delta[/latex]G = 0, so the following is true:
[latex]0=\Delta H^{\circ }-T\Delta S^{\circ }\qquad\text{ or }\qquad{T}=\dfrac{\Delta H^{\circ }}{\Delta S^{\circ }}[/latex]
Using the standard thermodynamic data from Standard Thermodynamic Properties for Selected Substances,
[latex]\begin{array}{rll}\hfill \Delta H^{\circ }&=&\Delta {H}_{\text{f}}^{\circ }\left(\ce{H2O}\left(g\right)\right)-\Delta {H}_{\text{f}}^{\circ }\left(\text{H2O}\left(l\right)\right)\hfill \\&=& -\text{241.82 kJ/mol}-\left(-\text{285.83 kJ/mol}\right)=\text{44.01 kJ/mol}\end{array}[/latex]
[latex]\begin{array}{rll}\hfill \Delta S^\circ &=& \Delta {S}^{\circ }\left(\ce{H2O}\left(g\right)\right)-\Delta {S}^{\circ }\left(\ce{H2O}\left(l\right)\right)\hfill \\ & =& \text{188.8 J/K}\cdot\text{mol}-\text{70.0 J/K}\cdot\text{mol}=\text{118.8 J/K}\cdot\text{mol}\hfill \end{array}[/latex]
[latex]T=\dfrac{\Delta H^{\circ }}{\Delta S^{\circ }}=\dfrac{44.01\times {10}^{3}\text{J/mol}}{118.8\text{J/K}\cdot\text{mol}}=370.5\text{K}=97.3^{\circ}\text{C}[/latex]
The accepted value for water’s normal boiling point is 373.2 K (100.0 °C), and so this calculation is in reasonable agreement. Note that the values for enthalpy and entropy changes data used were derived from standard data at 298 K (Standard Thermodynamic Properties for Selected Substances). If desired, you could obtain more accurate results by using enthalpy and entropy changes determined at (or at least closer to) the actual boiling point.
Check Your Learning
Key Concepts and Summary
Gibbs free energy (G) is a state function defined with regard to system quantities only and may be used to predict the spontaneity of a process. A negative value for [latex]\Delta[/latex]G indicates a spontaneous process; a positive [latex]\Delta[/latex]G indicates a nonspontaneous process; and a [latex]\Delta[/latex]G of zero indicates that the system is at equilibrium. A number of approaches to the computation of free energy changes are possible.
Key Equations
- [latex]\Delta G=\Delta H-T\Delta S[/latex]
- [latex]\Delta{G}=\Delta G^{\circ }+RT\text{ln}K[/latex]
- [latex]\Delta G^{\circ }=-RT\text{ln}K[/latex]
Try It
- A reactions has [latex]\Delta {H}_{298}^{\circ }[/latex] = 100 kJ/mol and [latex]\Delta {S}_{298}^{\circ }=\text{250 J/mol}\cdot\text{K}[/latex]. Is the reaction spontaneous at room temperature? If not, under what temperature conditions will it become spontaneous?
- Determine the normal boiling point (in kelvin) of dichloroethane, [latex]\ce{CH2Cl2}[/latex]. Find the actual boiling point using the Internet or some other source, and calculate the percent error in the temperature. Explain the differences, if any, between the two values.
- Without doing a numerical calculation, determine which of the following will reduce the free energy change for the reaction, that is, make it less positive or more negative, when the temperature is increased. Explain.
- [latex]\ce{N2}\left(g\right)+\ce{3H2}\left(g\right)\longrightarrow \ce{2NH3}\left(g\right)[/latex]
- [latex]\ce{HCl}\left(g\right)+\ce{NH3}\left(g\right)\longrightarrow \ce{NH4Cl}\left(s\right)[/latex]
- [latex]{\left(\ce{NH4}\right)}_{2}\ce{Cr2O7}\left(s\right)\longrightarrow \ce{Cr2O3}\left(s\right)+\ce{4H2O}\left(g\right)+\ce{N2}\left(g\right)[/latex]
- [latex]\ce{2Fe}\left(s\right)+\ce{3O2}\left(g\right)\longrightarrow \ce{Fe2O3}\left(s\right)[/latex]
Show Selected Solutions
1. [latex]\Delta {G}_{298}^{\circ }=\Delta {H}_{298}^{\circ }-T\Delta {S}_{298}^{\circ }[/latex]
[latex]\Delta {G}_{298}^{\circ }=100 - 298.15\times 250\frac{\text{J}}{\text{mol}\cdot\text{K}}\left(\frac{\text{1 kJ}}{\text{1000 J}}\right)=\text{25.5 kJ/mol}[/latex]
The [latex]\Delta {G}_{298}^{\circ }>0[/latex], so the reaction is nonspontaneous at room temperature.
- [latex]\Delta[/latex]G = [latex]\Delta[/latex]H − T[latex]\Delta[/latex]S
- [latex]0=100-T\left[250\frac{\text{J}}{\text{mol}\cdot\text{K}}\left(\frac{\text{1 kJ}}{\text{1000 J}}\right)\right][/latex]
- T = 400 K
Above 400 K, [latex]\Delta[/latex]G will become negative, and the reaction will become spontaneous.
Glossary
Gibbs free energy change (G): thermodynamic property defined in terms of system enthalpy and entropy; all spontaneous processes involve a decrease in G
standard free energy change ([latex]\Delta[/latex]G°): change in free energy for a process occurring under standard conditions (1 bar pressure for gases, 1 M concentration for solutions)
standard free energy of formation [latex]\left(\Delta {G}_{\text{f}}^{\circ }\right)[/latex]: change in free energy accompanying the formation of one mole of substance from its elements in their standard states
Licenses and Attributions (Click to expand)
CC licensed content, Shared previously
- Chemistry 2e. Provided by: OpenStax. Located at: https://openstax.org/. License: CC BY: Attribution. License Terms: Access for free at
https://openstax.org/books/chemistry-2e/pages/1-introduction
thermodynamic property defined in terms of system enthalpy and entropy; all spontaneous processes involve a decrease in G
change in free energy for a process occurring under standard conditions (1 bar pressure for gases, 1 M concentration for solutions)
change in free energy accompanying the formation of one mole of substance from its elements in their standard states
